Closing the loop on genome folding
Researchers recorded the folding dynamics of the human genome over time, creating a novel 4-D map of genome organization.
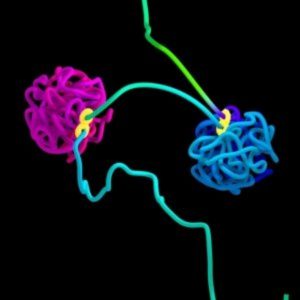
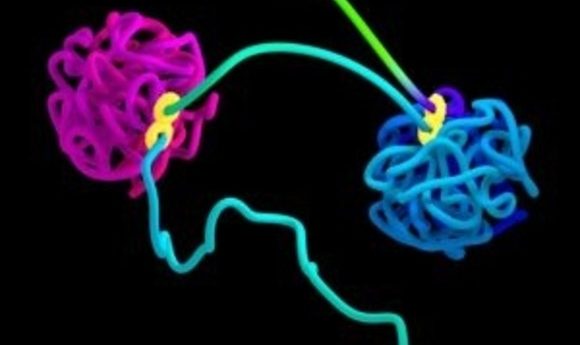
Simulation of DNA organized into two globules by loop extrusion. The figure-8 structure indicates the extrusion complex.
Credit: Najeeb Marc Tarazi, Adrian Sanborn, Erez Lieberman Aiden
We are all familiar with the hopeless tangling of headphone cords within minutes of storing them in a pocket. But just imagine stretching out a meter long string and then packaging it into a space a million times smaller, without introducing a single knot. This may sound like a magic trick, but literally every cell in your body has achieved this feat.
Not only is the human genome tightly packed into the tiny nucleus of the cell, but it regularly unfolds efficiently during gene expression and replication. This hints that genome folding is a non-random process, carefully orchestrated to avoid catastrophic events resulting from a tangled mess of DNA.
Apart from solving an amazing packaging problem, genome folding is also intimately connected with gene expression. Gene functions are influenced by activator and repressor proteins contacting select regions on the DNA, but these binding sites may be physically located far apart on the genome. For gene expression, they are brought together by the formation of DNA loops.
Not knowing the elusive elements that control loop formation has hindered a detailed understanding of genome folding for decades. Now, in a new study published in Cell, researchers mapped folding dynamics of the human genome over time by modulating levels of a protein, thus confirming its essential role in the process (1). While 3-D genome maps existed , this novel 4-D map may provide new insights into understanding the processes involved in genome folding and organization.
Keeping DNA in the Loop
“The genome is not just a string of letters but is rather a physical entity,” said Suhas Rao from Stanford University School of Medicine, first author of the new study. When the genome folds, the resulting contacts between its different regions have functional consequences in modulating gene expression.
To better understand genome folding, Suhas Rao and colleagues mapped DNA loops in 3-D a few years ago (2). Fluorescence microscopy is a convenient tool for gaining spatial information regarding a few DNA fragments by labeling them with fluorophores, but it is impractical to apply this method on a genome-wide scale. So, Rao’s team used a special method, Hi-C, for capturing the genome conformation.
To perform Hi-C, the researchers first crosslinked the chromatin in cells from a human cell line and then digested the DNA using restriction enzymes. They marked the ends of the digested DNA with biotin prior to ligation to ensure that only ends close to one another ligated together. Then, the team cut the DNA into millions of pieces and sequenced the fragments. The resulting map of spatial organization of chromatin showed two types of domains: loops and compartments.
Loops are formed by pinching local DNA sections together via protein anchors, called an extrusin complex. “Think about it like the strap adjustor on your backpack. The strap adjustor has two pieces, and then you can fold the strap through and make a bigger and bigger loop,” explained Rao. The compartmentalization process, on the other hand, brings distant parts of the genome into contact.
From 3-D to 4-D
Although the previous study revealed 3-D genome-folding patterns, this was merely a snapshot of the genome structure averaged from millions of cells. Rao was curious about the temporal dynamics of genome folding. However, a time-course experiment requires synchronizing the cells by eliminating the extrusion complex machinery. This is not simple.
In the past, several studies predicted that all loop domains should disappear upon destroying cohesin, an integral component of the extrusion complex. But when researchers tried to deplete cohesin or its related proteins, they found little effect on the loops, contrary to their expectations. Rao believes this might be due to effects of remnant protein in the cell.
In their new study, Rao and his team used a powerful tool to completely get rid of cohesin. They fused a core protein of the cohesin complex with a tag that degraded the protein in the presence of an externally added plant hormone from the auxin class, which in this case simply served as a trigger. Moreover, the system was reversible: eliminating the auxin restored cohesin in the cell. Using microscopy, the team confirmed that 95% of cohesin proteins were eliminated from the cell when triggered with the auxin, and these were restored when the auxin was withdrawn. Both processes occurred within a short span of 40 minutes. Importantly, the researchers also found that all loop domains disappeared when cohesin was destroyed, and they reappeared upon replenishing the cohesin. By combining this sophisticated cohesin degradation system with Hi-C, the team successfully added the fourth dimension—time—to their genome-folding maps.
“This is a really, really important paper,” said Anders Sejr Hansen from the University of Berkeley, who was not involved in this study. “It establishes, in a completely unambiguous manner, that loop domains require cohesin.”
Xavier Darzacq from the University of Berkeley, who also was not a part of this work, seconded this opinion. “Now that we have these experiments, we can claim a causal effect, which in biology these days is a rare commodity.”
Compartmentalization without Cohesin
Along with confirming the role of cohesin in extrusion, Rao made some surprising observations. First of all, loss of cohesin had no effect on the transcription of approximately 80% of the genes. This was astonishing because the researchers assumed that loop domains were critical in regulating gene transcription. Although loop domains seem to only affect a minority of genes, their influence on downstream gene expression could be significant if the affected genes were regulatory genes.
Secondly, destroying cohesin strengthened, rather than destroyed, the compartmentalization loops. These findings agreed with another study that reported extrusion and compartmentalization as independent processes (3). While proteins are crucial for extrusion, compartmentalization is governed by interactions between regions bearing similar histone modifications, such as acetylation, ubiquitinylation, and methylation.
To consolidate their experimental data with simulations, the team collaborated with Tamar Schlick, a modeling expert at New York University Schlick modeled a DNA fiber with alternating acetylated and non-acetylated parts, treating the nucleosome with the wrapped DNA as an electrostatically charged object.
In her model, Schlick noted an intrinsic tendency of the DNA fiber to segregate where two modified histone tail segments come together or two unmodified regions come together, just like in the experiments. Schlick was not surprised. “Clearly the proteins are very important, but they have to know when and where to bind, and that binding has to be dictated by structure. The intrinsic signatures of folding have to come from the DNA itself,” she said.
Rao believes that the study details the underlying mechanisms behind ideas that have been around for long time, demonstrating their combined role in shaping transcription. “There is all this wealth of ways to bring small regions of the genome that are separated by vast spatial differences and genomic differences together very quickly, which I think is really exciting,” said Rao.