Chasing Zika virus
A year ago, Zika infections were reported in southern Florida for the first time, marking the virus’ arrival in the continental United States. How did it get there, and what can be learned to prevent future outbreaks?
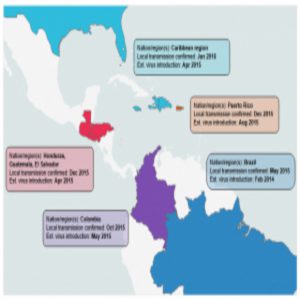
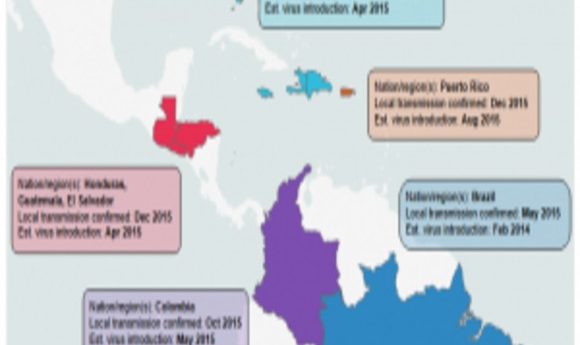
By analyzing genomes generated as part of the study, researchers confirmed that the Zika virus was present in affected countries well before the first confirmed cases were detected. Credit: Tom Ulrich, Broad Institute Communications.
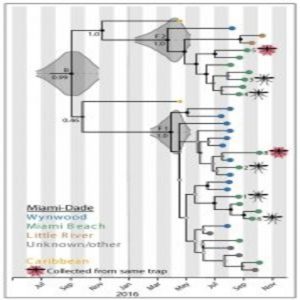
Viral genome sequencing revealed multiple introductions of Zika virus to Miami from the Caribbean.
It’s said knowledge and understanding can conquer almost any fear. For Zika virus, however, fear seemed almost insurmountable when infections started to appear in the Miami area during the summer of 2016. Although there were more than 200 confirmed infections, Zika did not turn into the major epidemic in the Southern US that some had predicted, thanks to a more temperate climate, the limited range of the mosquito vector, Aedes aegypti, and modern sanitation. However, the possibility of the virus returning in a more transmissible form remains, as well as the possibility that other flaviviruses transmitted by the same mosquito, such as Dengue and Yellow fever, could be spread.
Thankfully, a large team of researchers from multiple institutions in the US, Brazil, and other nations have now analyzed the evolution and transmission of Zika virus at the molecular level, providing a powerful weapon against future outbreaks (1–3).
Scarce Starting Material
The very nature of Zika infections makes the virus difficult to research. “Relative to other viral infections, Zika virus infections are often very low titer—peaking quickly and dropping off quickly,” noted Bronwyn MacInnis of the Broad Institute, one of the research team leaders. “Sequencing to detect Zika RNA from patient samples was like looking for a needle in a haystack… On both the lab and the computational end, we had to develop new methods to tackle this challenge.”
Jason Ladner of the US Army Medical Research Institute of Infectious Diseases (USAMRIID), lead author of a paper reporting analysis of the Florida infections (2), also noted the difficulties of obtaining high-quality Zika sequence: “Another challenge was a lack of [sequence] data from other Zika cases outside the US due to sparse sampling and a lack of whole-genome sequence.”
Despite this, MacInnis, Ladner, and their teams were able to get a handle on Zika thanks to some innovative sequencing techniques used to study the spread of Zika in Brazil (1). In the Brazilian study, Nuno Faria and colleagues took advantage of a new multiplex PCR technique optimized for either the MinION portable sequencing platform or Illumina technologies (4) to rapidly identify Zika in blood, urine, and serum samples from infected patients. This protocol works well for low-abundance RNA virus genomes, where standard metagenomic sequencing techniques are often insufficient. But in the end, finding the best approach was a team effort.
“We and our colleagues each took a unique approach to cracking the nut of Zika sequencing. We developed hybrid capture for Ebola sequencing and combined this with unbiased metagenomic sequencing and multiplex amplicon PCR sequencing,” MacInnis explained. “Comparing the different approaches allowed us to determine the quality of the protocols and the data they generated.”
Ladner’s group used the multiplex PCR sequencing approach and probe-based targeted enrichment sequencing methods to detect Zika virus in mosquitos and patient samples (3). His team found urine to be the best sample from which to obtain consistent results, due to its relatively high viral titer. “It had a higher concentration at later timepoints of infection. Whole blood would likely also be better than serum at these later points, but we didn’t have many whole blood samples available from Florida.”
Tracing the Spread of Zika in the Americas
With technical details worked out, the researchers started to analyze the spread and evolution of Zika in Florida (2) and the Americas outside of Brazil (3) by sequencing both patient and mosquito samples in order to determine the most recent common ancestral strain of the virus and thus establish where the virus originated and its geographic transmission pattern. “Like many other viruses, Zika evolves quickly and in a more or less regular clock-like manner. Therefore, we can fit the genetic data to a molecular clock to determine when the virus was likely introduced. We can also use the genetic sequences to estimate the number of different introductions.”
Faria’s group sampled patients from clinics throughout Brazil and applied molecular clock analysis to the sequence data to show that Zika arrived in northeast Brazil sometime in late 2013 or early 2014, almost a year before infections appeared.
The other research teams ascertained that the majority of the Zika viruses present in Florida were most closely related to viruses from the Caribbean. In fact, they determined that there were at least three separate introductions from the Caribbean into the Miami area. Gytis Dudas, a postdoctoral fellow at the Fred Hutchison Cancer Research Center and co-author of the study on Zika transmission in Florida (2), was initially surprised by the finding. “[The Caribbean] is not a particularly populous region of the world compared to Brazil, where Zika first established itself,” he said. “Once we had the incidence data, however, it became obvious that despite not having many cases overall, the Caribbean on average had more cases per capita. We convinced ourselves of this pattern even further when we looked at when and in what numbers people travel to the Caribbean from Florida.”
Although Zika was first identified in Uganda in the late 1940s, scientists knew very little about the virus until recently. “The most surprising thing we learned was how the virus is introduced in different regions,” MacInnis said. “There was a single introduction into Brazil and early introductions into other places. The observation that the virus circulates much earlier than expected helps us understand how the virus unfolds and how this might correlate with clinical symptoms. The data we have collected will serve as a resource for developing Zika therapies and preventing future outbreaks, for example by helping to distinguish the source of new infections, which is helpful in guiding the public health.”