Sequencing DNA methylation

It’s time to turn to sequencing the epigenome, the DNA modifications that alter gene expression and determine our individual genetic identity.

DNA molecule with methylation on the center cytosine bases. Image courtesy of Christoph Bock, MPI-INF.
Epigenetics is the study of changes in gene expression that occur without any changes in DNA base sequence, resulting in a change of phenotype without affecting the genotype. Whilst all cells have the same DNA, the epigenetic patterns differ, allowing for a greater variety of phenotypes in the population.
Sequencing the genome may not be enough in determining an individual’s genetic make-up as it is not just the DNA-coded proteins that cause differentiation. Therefore, to truly understand the genome it is necessary to also sequence the epigenome.
DNA methylation
There are three methods of epigenetic changes; histone modification, chromatin remodeling and, the most common, DNA methylation (DNAme).
In eukaryotes, DNAme occurs when a DNA methyltransferase enzyme adds a methyl group to position 5 of the ring of a cytosine nucleotide. The cytosine is usually in a CpG dinucleotide pairing; therefore, methylation forms methyl-CpG (5mC).
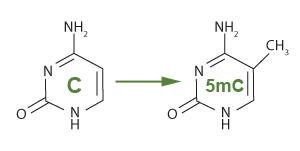
The methylation of cytosine using DNA methyltransferase to add a methyl group (CH3)
Rather than turning genes off, DNAme acts by preventing their activation. Promotor regions of genes are often particularly highly methylated, and the silencing of genes may in part be due to this [1]. This methylation can inhibit gene expression via two mechanisms; modifying the cytosine base to inhibit DNA binding factors or activating 5mC binding proteins [2].
These proteins recruit factors that form a closed chromatin structure thus making the gene less accessible to transcription factors and preventing genetic replication. The covalent attachment of the methyl group means that once formed the methylation is chemically stable, making it a strong structural foundation for gene expression in differentiated cells [3].
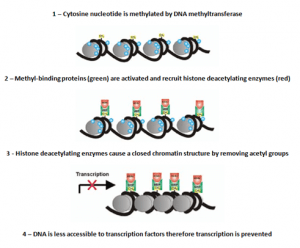
The process of silencing gene expression due to DNA methylation (Adapted from [4])
DNAme has vital roles in embryonic development and maintenance of cell pluripotency [5]. These roles may occur spontaneously, be inherited or be influenced by the pre-natal environment. Differing environmental factors allow the epigenome to change throughout life, causing phenotypic plasticity.
Sometimes these alterations are due to negative life events, such as stress. Therefore, changes are usually negative and can be linked with behavioral instability, premature aging and various diseases.
There are two key groups of methods currently used for DNAme sequencing; those involving bisulfite conversion and those involving methylation enrichment.
Bisulfite conversion sequencing
Use of standard PCR amplification techniques erases methylation patterns so cannot differentiate methylated or un-methylated cytosine bases. Therefore, it is necessary to perform extra steps in order to maintain the required methylation information. Bisulfite conversion involves targeting adaptor-tagged DNA templates and converting the cytosine bases to uracil whilst leaving the 5mC intact. During standard PCR amplification, uracil will be recognized as thymine and the 5mC is recognized as cytosine [6]. Therefore, although the methylation state is erased, it can be inferred that any cytosine nucleotides shown in the analysis were originally methylated.
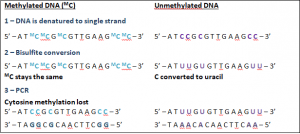
The effect of bisulfite conversion of methylated vs unmethylated DNA.
Following the conversion, there are two methods for sequencing other than traditional PCR; MethylC-sequencing (methylC-seq) or reduced representation bisulfite sequencing (RRBS).
Whole-genome bisulphate methylC-seq (WGBS) is the most comprehensive method for DNAme sequencing, providing single-base methylation profiles for more than 90% of the CpGs in the genome. The primer design needed for the traditional PCR methods can often introduce bias; using cytosine-methylated universal adaptors makes targeted primer design unnecessary therefore the likelihood of bias is reduced.
MethylC-seq is also able to detect features of DNAme not readily picked up by traditional methods, such as identification of non-CpG methylation sites [6]. However, this technique requires micrograms of input DNA, needs multiple rounds of PCR amplification and is expensive to run, particularly for large-sized genomes.
A more commonly used, cost-effective method is RRBS. This method sequences a shorter, representative fraction of the genome and reviews multiple cytosine positions within multiple fragments. Choice of RRBS loci could be beneficial for determining the function of different genomic regions and how they can be epigenetically altered. However, although RRBS may improve resolution in marker-based approaches, in species that lack a good reference genome there are limitations in the functional conclusions that it can draw [7].
A key limitation of using a bisulfite sequencing method is the chance of incomplete conversion. Determining the methylation sequence requires all unmethylated cytosines to be converted to uracil; any unconverted cytosine nucleotides would result in false positives [8]. The bisulfite conversion process also raises issues as the necessary conditions may damage the DNA. High temperatures and long incubation times can lead to approximately 90% DNA degradation [9].
Methylation enrichment sequencing
Methylated DNA immunoprecipitation sequencing (MeDIP-seq) and methylated DNA binding domain sequencing (MBD-seq) both act by enriching the methylated regions before sequencing. When used alone these avoid the potentially damaging effects of bisulfite.
MeDIP-sequencing involves fragmenting the DNA into various lengths and then using a 5mC antibody to target the methylated fragments. Although this is a relatively low-cost method that can sequence the whole-genome DNA, it has poor resolution, which makes it difficult to precisely identify the methylated sites. A way to improve the MeDIP-seq efficiency could be to combine it with WGBS to create a high-resolution, low-cost sequencing method [10].
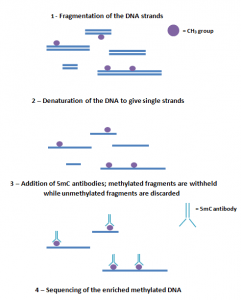
The process of MeDIP sequencing
MBD-seq uses the MBD2 protein to enrich methylated double-strand DNA fragments [5]. Affinity of the protein to the binding domain is modulated by ionic strength with changes relative to the degree of methylation. Because of this, MBD-seq often favors regions of high CpG density and may miss lower density regions [11]. To identify all regions, combination methods are likely the most effective technique.
Although both methods have benefits, it is in combination with bisulfite conversion where they have the greatest efficacy. Integration methods may be beneficial for reducing costs whilst maintaining resolution and high genomic coverage.
Uses for epigenetic sequencing
Epigenetic sequencing has multiple potential applications due to its increased specificity to an individual over and above traditional genome sequencing, even allowing for the differentiation of monozygotic twins. Such applications include detecting environmental risk factors, acting as disease biomarkers or predicting treatment outcomes.
Epigenetic changes that occur throughout life can affect aging and may alter an individual’s predisposition to neurological or autoimmune disorders. This methyl aging clock differs from chronological age and DNAme sequencing may be able to predict age of death; often those who live longer have a younger epigenetic age compared with those who die young [3].
Sequencing DNAme may also be able to predict risk of cancer as it is suggested that cancer is positively correlated to the degree of abnormal age-related methylation [12]. Epigenetic dysregulation likely plays a role in tumor development therefore DNAme sequencing may have a role in cancer diagnosis [13]. Testing methylation status of tumor cell biopsies could help to distinguish the primary source of any cancer, particularly important if only detected at late stages of metastasis. Recent studies have demonstrated the use of DNAme sequencing in determining glioblastoma disease progression and sub-type classification [14], something that could have a significant impact on the development of personalized medicine.
The brain has the highest level of DNAme of any tissue in the body; therefore, methylation can have a strong influence on behavior and mental health [15]. Altered DNAme has been found in certain genes for depression in humans such as the brain derived neurotrophic factor (BDNF) gene promotor, whose methylation status has been suggested to predict response to treatment and therefore, if sequenced, may assist in personalized medicine [16].
Future for the field
Advances in technology allow potential for clearer results, reduced limitations and novel applications. One such advancement is post-bisulfite adaptor tagging (PBAT); here the bisulfate treatment occurs before the DNA is tagged with adaptors therefore preventing DNA degradation and reducing the amount of DNA that is needed in the sample. It removes the need for PCR amplification, making it a valuable method for WGBS [17].
PBAT may also be used to improve the efficiency of single-cell bisulfite sequencing, although in this situation PCR is required [18]. Bisulfite conversion methods may soon be obsolete with the development of methods such as single molecule, real-time sequencing; DNA modifications are detected using the pausing time of the polymerase enzyme when it encounters a modified base [19] thereby removing the need for pre-sequencing sample preparation.
CRISPR/Cas9 technology is an important advance in the field of genetics and its editing methods may be applied to the epigenome. Primarily, this may be used to give greater clarification regarding the relationship between methylation and cell behavior and function; information that may later be translated to the clinic. For editing DNAme it is proposed that the Cas9 enzyme interacts with the DNA methyltransferase to increase methylation at CpG sites [20]. This could be used in differentiating stem cells, artificially creating cells needed for transplants.
Although often overlooked relative to the genome, the epigenome may be equally if not more important in determining an individual’s genetic make-up. With further research DNAme could have potential to become as common place as genome sequencing in determining genetic identity.