How to: quantitative proteomics with stable isotope standard protein epitope signature tags

Proteomics is a research field focusing on the large-scale study of proteins in biological contexts. The goal is to describe various biological questions related to aging, drug treatment, disease, and recovery by analyzing protein changes in a comprehensive and quantitative manner [1].
Proteins can be studied in a biological specimen of different origins using various techniques. Typically, bodily fluids; such as blood, its liquid part blood plasma, saliva, urine, cerebrospinal fluid; tissues and cell lines are analyzed by mass spectrometry (MS) [2], using affinity reagents [3], or even by combining these two technologies [4]. All human sample types, especially blood plasma, hide an immense amount of information about an individual’s physiological state. Blood plasma contains a proteome that is by far the most complex with a vast dynamic range of protein concentrations, which are of different origins and functions, covering the whole repertoire of human proteins. It is therefore a great sample type to collect and analyze in relation to health and disease, but also the most difficult to explore.
The most widely used technique for proteomics analysis is an online system of liquid chromatography (LC) separation coupled with mass spectrometry (LC-MS). MS is an analytical technique that one can imagine as an extremely sensitive scale. It is capable of measuring the mass of single ions together with their charge and the results are presented in a mass spectrum with the intensity as a function of the mass-to-charge ratio (m/z). The LC functions as a separation step prior to the MS analysis and adds another dimension to the spectra: time.
Contents:
Targeted Proteomics
Targeted proteomics is an approach within bottom-up proteomics meaning it uses products of enzymatic digestion (peptides) to infer protein quantities. It is an excellent tool for performing measurements with great reproducibility and sensitivity making it a suitable method for application in a clinical setup. In contrast to the widely used data-dependent acquisition (DDA), also referred to as shotgun proteomics, targeted proteomics works with a predefined set of peptides and builds on previous knowledge about the sample.
In a bottom-up proteomics experiment, only peptides that are unique to one protein are used for quantification in order to infer protein abundance in a sample. The accuracy and especially the precision of quantification are crucial for finding differences in protein changes as a result of biological effects. The instrumentation for generating quantitative data in targeted proteomics experiments includes a variety of mass detectors and configurations of MS.
There are three main types of MS instruments that are used for targeted proteomics approaches: orbitrap, triple quadrupole (QQQ) and quadrupole time of flight (qTOF). They differ in the way of navigating and analyzing ions inside the instrument, and each one of them have their niche in the proteomics field, which inherently comes with different modes of MS operation. The selective reaction monitoring (SRM) and multiple reaction monitoring (MRM) are modes used on QQQ and qTOF instruments whilst parallel reaction monitoring (PRM) is used on orbitraps. Each instrument and method have pros with QQQ being the most sensitive, qTOF the fastest and orbitrap the best in resolution, allowing it to distinguish between two ions with very similar m/z. But they each come with cons where QQQ is the most laboursome for method development, qTOF instruments are large and generate extremely big files and orbitraps are expensive and slow in recording m/z of analytes.
A special place within targeted proteomics is held by the data-independent acquisition (DIA), which connects the fields of targeted and exploratory proteomics. Here, all the analytes are sequentially analyzed in groups of bigger m/z windows. The targeted part of the analysis lays in the processing of the data and the targeted extraction of preselected peptide chromatograms. The DIA is typically performed on either orbitrap or qTOF instruments and is superior in the unbiased way of collecting the data and the option for re-mining the data for new targets but requires rigorous data processing workflows.
Quantification
In targeted experiments, the area under the curve (AUC) from extracted ion chromatograms is typically used as a measurement and can be interpreted in several ways as a quantitative assessment of peptides. In general, the quantification of a target peptide is either relative or absolute. Relative quantification is a measure where the amount of an analyte is described in relation to another measurement of the same analyte across multiple biological samples or between two groups, such as in case-control studies.
Absolute quantification is the determination of the abundance of analyte on the absolute scale, typically mg/ml or pmol/ml. This can be done using stable isotope labelled (SIL) heavy standards. SIL standards contain 13C and 15N enriched arginine and lysine and typically cover shorter amino acid sequences or whole sequences of endogenous proteins. They are spiked-in to samples in known concentrations allowing for an independent quantification. In practice, chromatograms from the endogenous analyte and corresponding SIL standard are overlayed resulting in a ratio of heavy (SIL) and light (endogenous) AUCs. AUC of the quantitative target peptide is linearly related to its amount and, therefore, the absolute amount of the analyte can be calculated from the known amount of spiked-in SIL standard (Figure 1).
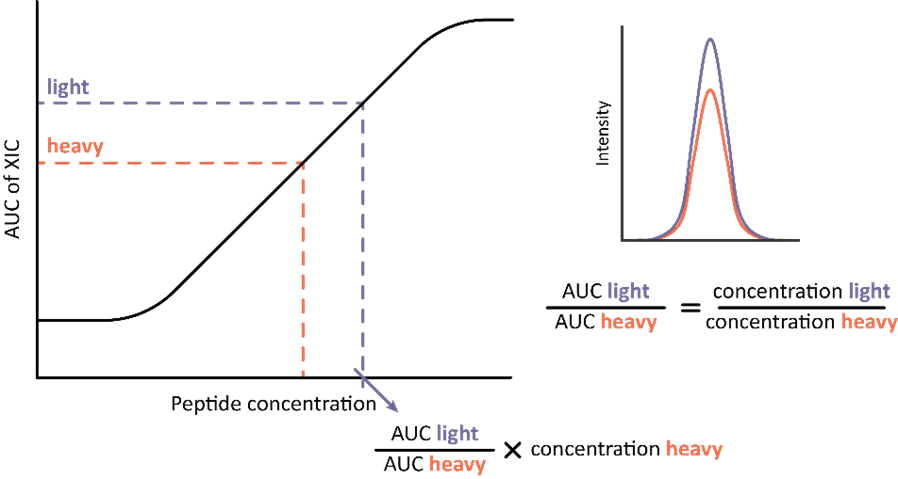
Figure 1: Workflow for absolute quantification based on extracted ion chromatograms (XIC) of heavy and light versions of the same peptide. The peak areas are overlayed, and their ratio calculated. The concentration of endogenous peptide is calculated by multiplying known spike-in concentration of the heavy standard with the ratio of corresponding peak areas.
To be able to detect the smallest differences in protein levels between different sample cohorts, a well-developed robust workflow is crucial. This includes a well-defined and experimentally tested digestion protocol together with a calibrated and well-performing LC-MS setup as the foundation. This should be combined with a stringent targeted proteomics assay development pipeline and reproducible data processing. Furthermore, including heavy-labelled standards in the sample preparation can greatly improve the reproducibility and precision, especially when using state-of-the-art protein standards, which also allow for absolute protein quantification. Stable isotope standard protein epitope signature tags (SIS-PrESTs) [5] have a great advantage for being relatively simple to produce and easy to work with since they cover short 50-200 amino acid stretches of proteins (Figure 2).

Figure 2: Amino acid sequence alignment of the SIS-PrEST and its corresponding endogenous protein with their tryptic peptides.
They can be utilized in all different MS setups and analysis modes including both targeted (SRM, MRM, PRM, DIA) and untargeted (DDA) modes of MS operation. They are added to the sample as a first step of the proteomics workflow and also mimic the digestion efficiency of the endogenous protein. This makes them a robust choice for absolute quantification since all the bias from the downstream sample preparation is accounted for, such as tryptic digestion.
SIS-PrEST workflow
Target selection
Since each SIS-PrEST standard is a polypeptide covering a single endogenous protein, they are added as the very first step of sample analysis workflow prior to enzymatic digestion or other sample processing. Therefore, all the protein targets that are needed to be quantified have to be selected prior to the sample processing and analysis. It is usually done during an iterative process where only few representative samples are selected for method development and screened using either DIA or DDA. These initial experimental results are used to generate a list of proteins that could have been identified and later can be compared with available SIS-PrEST sequences. When an overlap is found it can be selected for subsequent quantification. Currently, there are 3,400 SIS-PrESTs covering over 2,100 protein targets.
On the other hand, SIS-PrESTs can be used to generate MS coordinates in SRM, MRM or PRM mode. Here, a handful of SIS-PrESTs are digested and used for screening all of the theoretical peptides, which are then verified in a sample of choice. Such a process is rather laboursome but since these modes of MS operations are more sensitive, more targets can be included in the final quantitative assay.
Titration to endogenous levels
When the targets of interest are known, one has to add them to each sample in close to a 1:1 ratio with the endogenous protein. This is done in an iterative process or by constructing a calibration curve. Endogenous proteins are measured using a one-point calibration strategy optimized for high quantification precision. This is achieved by adjusting the spike in of the SIS-PrESTs levels to be as similar to endogenous levels as possible.
Pooling
A great advantage of SIS-PrESTs is that they are individually produced, purified, and absolutely quantified. Therefore, it is possible to generate custom-made pools for multiplexed absolute quantification. One just needs to make a calculation of how many samples there are to be analyzed and combine as much of each SIS-PrEST as needed, based on the titration experiments. Such a pool can be added to samples directly but can also be aliquoted into a 96-well plate, for example, and vacuum dried. Such plates can be generated in high throughput and stored at room temperature for up to 1 month, sent between the labs in a regular mail or stored at -20 °C. Having all the plates prepared in a single batch can further increase the precision of analysis, and it also facilitates further sample preparation, especially when processing large cohorts [6].
Sample preparation
A standard proteomics workflow includes reduction, alkylation, and enzymatic digestion. This can be done under different buffer conditions and using different reagents. The main focus should be to test a few protocols from the literature and decide on a single one that is used for at least a whole study. A major benefit of using vacuum-dried standards is the ability to just take a plate out from a freezer and add the sample, which is followed by all the reagents needed. Such workflow setup allows for addition-only protocols where all reagents are added into the same reaction container. This setup assures the best analytical precision.
About the author:
 David Kotol (ProteomEdge, Stockholm, Sweeden) holds a master’s degree in biotechnology from the University of Chemistry and Technology (Prague, Czech Republic), after which he earned a PhD in biotechnology from the KTH Royal Institute of Technology (Stockholm, Sweden). David’s research focuses on absolute quantification of proteins in human blood plasma and he is the first author of a paper published in BioTechniques on this topic.
David Kotol (ProteomEdge, Stockholm, Sweeden) holds a master’s degree in biotechnology from the University of Chemistry and Technology (Prague, Czech Republic), after which he earned a PhD in biotechnology from the KTH Royal Institute of Technology (Stockholm, Sweden). David’s research focuses on absolute quantification of proteins in human blood plasma and he is the first author of a paper published in BioTechniques on this topic.
- Deutsch EW, Omenn GS, Sun Z et al. Advances and utility of the human plasma proteome. J. Proteome Res. 20(12), 5241-5263 (2021).
- Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D and Mann M. Plasma proteome profiling to assess human health and disease. Cell Syst. 2(3), 185-195 (2016).
- Thul PJ, Åkesson L, Wiking M et al. A subcellular map of the human proteome. Science 356(6340) eaal3321 (2017).
- Schwenk JM, Gry M, Rimini R, Uhlén M and Nilsson P. Antibody suspension bead arrays within serum proteomics. J. Proteome Res. 7(8) 3168-3179 (2008).
- Gold L, Ayers D, Bertino J et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. Nature Precedings 1-1 (2010).
- Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW and Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 3(2) 235-244 (2004).
- Anderson NL and Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell. Proteomics 1(11) 845-867 (2002).
- Sajic T, Liu Y and Aebersold R. Using data-independent, high-resolution mass spectrometry in protein biomarker research: perspectives and clinical applications. Proteomics Clin. Appl. 9(3-4) 307-321 (2015).
- Picotti P and Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 9(6) 555-566 (2012).
- Liebler DC and Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry 52(22) 3797-3806 (2013).
- Olsen JV, Ong S-E and Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3(6) 608-614 (2004).
- Peterson AC, Russell JD, Bailey DJ, Westphall MS and Coon JJ. Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11(11) 1475-1488 (2012).
- Gillet LC, Navarro P, Tate S et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11(6) (2012).
- Ong S-E and Mann M. Mass spectrometry–based proteomics turns quantitative. Nat. Chem. Biol. 1(5) 252-262 (2005).
- Lesur A, Schmit P-O, Bernardin F et al. Highly multiplexed targeted proteomics acquisition on a TIMS-QTOF. Anal. Chem. 93(3) 1383-1392 (2021).