Prying into prion structure
Antibody-based immunotherapy has recently emerged as a potential treatment for prion-mediated neurodegenerative diseases. But is this approach entirely safe for patients?
In the 35 years since Stanley Prusiner coined the term prions to describe infectious proteins that cause transmissible spongiform encephelapathy (TSE), the victims of this progressive and fatal neurodegenerative disorder have known little relief. In recent years, immunotherapy appeared as a promising approach to treat this hitherto incurable disease by using antibodies to target the misfolded prion proteins that kill brain cells. Now, a new study from Bei Wu and colleagues at Boston University demonstrates that certain prion-targeting antibodies can have toxic effects on neurons.
“It’s a very interesting study that seems to tie together various strands of science that have been dealt with rather separately in the literature till now, but which this study sort of replicates and extends and tries to draw together as a mechanistic contribution towards the way that the prion protein may be involved in modulating cell survival and cell death,” said Andrew Gill of the Roslin Institute, University of Edinburgh, who was not associated with the study.
Similar to many other neurodegenerative diseases, prion diseases are caused by misfolded proteins aggregating in the brain and causing neurons to die. Unlike other neurodegenerative diseases, however, the misfolded prion protein acts as a template for the rapid generation of more misfolded proteins, and in rare cases, can be transmitted between individuals. The normal, correctly folded form of one prion protein, PrP, which is encoded by the Prnp gene, is expressed in high levels in the brains of all individuals, and figuring out its physiological function has been a major focus for researchers studying such diseases.
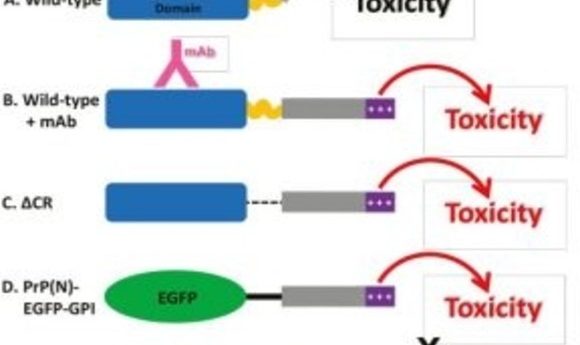
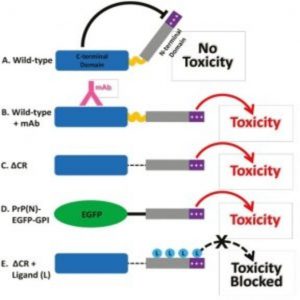
PrP has two main domains: the N-terminal domain is flexible, while the C-terminal domain is more structured and rigid. Previous studies showed that deleting a small central region connecting the two domains leads to toxicity in neurons, accompanied by the generation of spontaneous ionic currents, which the study authors suspect may be one of the physiological functions of PrP..
Interestingly, when the researchers blocked certain portions of the PrP N-terminus using antibodies or specific ligands, these currents disappeared. Because of this, the authors wondered if the N-terminus could be responsible for causing neuronal toxicity. In support of this hypothesis, the researchers found that the N-terminal domain, when expressed alone in cells without the C- terminal domain, gave rise to spontaneous ionic currents, which could be blocked by antibodies and ligands directed to various parts of the N-terminal domain.
The observation that may have the most impact on the field of immunotherapy for prion disease, however, was that similar spontaneous currents were also generated by antibodies that bound an alpha-helical region in the C-terminal domain of PrP. The authors found that when they added these C-terminal-targeted antibodies to cultured hippocampal neurons, the neurons adopted an abnormal form of dendritic morphology called “beading,” a marker of cellular toxicity. Blocking the N-terminal domain using antibodies or ligands, on the other hand, eliminated this toxic effect, as well as the spontaneous currents.
The authors hypothesize that the N-terminal domain of PrP is capable of adversely affecting brain cells and that this toxic effect is normally kept in check by the C-terminal domain. When antibodies against the C-terminal domain block this interaction, the toxic effects of the N-terminal domain are unmasked, leading to dendritic abnormalities.
“My research will remind people to be careful to use the antibody, at least to be careful with antibodies targeted to the C-terminus domain, especially α-helix 1 domain of prion protein,” said Wu. She and her colleagues are currently working on understanding the link between the induced spontaneous currents and neuronal toxicity observed in this model.