Modern microscopy: live-cell imaging with super-resolution
As the super-resolution microscopy industry evolves, experimental needs and demands change alongside it. More and more, demand for super-resolution microscopy on live samples has grown and caught the attention of microscope manufacturers. In this article, we will discuss practical limitations for live cell imaging with super-resolution in as well as methods to mitigate their effects on experimental data.
Lauren Alvarenga is a product manager for research imaging as part of the Scientific Solutions Group within the Olympus Corporation of the Americas. She is currently responsible for imaging software, inverted and super-resolution microscopes. Lauren received her BSc in biomedical photographic communications from the Rochester Institute of Technology (NY, USA).
Super-resolution has been an important topic within the microscopy community since the 2014 Nobel Prize in Chemistry. In spite of efforts to simplify and perfect the process of breaking the diffraction limit, it is well known that super-resolution is difficult to perform in practice, even more so with live samples. This article highlights various super-resolution methods and techniques that can help researchers get the most out of their live cell experiments.
Two of the more common super-resolution methods are techniques that are usually grouped together called ‘localization microscopy.’ With localization methods, such as PALM and STORM, photoswitchable fluorophores are used to excite only a small number of molecules at a time. Since these spots are still technically diffraction limited, information from their theoretical point spread function is used to fit them down to a range of 10–20 nanometers. This is repeated over and over again until an image is formed.
However, acquisition for localization methods can require thousands of frames, which can take a very long time. Performing this frequent switching can also present some practical issues, as specific fluorophores and imaging buffers are sometimes required.

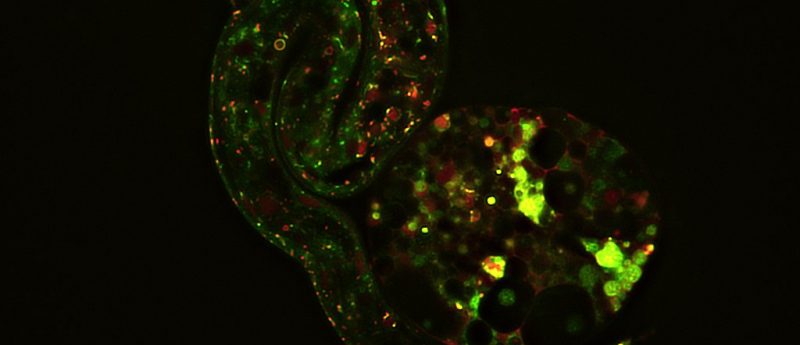
Figure 1. Super-resolution techniques, enabled by software and hardware advances, highlight the most minute details and processes that define life. An example is this image depicting early embryonic development captured on the Olympus SpinSR super-resolution microscope. (Image courtesy of Dan Houchin, Olympus America.)
Choosing a super-resolution method
Some techniques specifically focus on fluorescent proteins, which can be easily worked into existing transfection protocols. New fluorophores are coming out with increasing frequency, and tools like FPVIS help visualize how current fluorophores compare against other popular choices—choosing the right fluorophore is critical to live cell experiments. Hardware advances have also been beneficial for localization microscopy. Real-time controllers reduce overhead time for image acquisition, which can increase speed and accuracy during fast paced switching.
Structured illumination, also known as SIM, is another popular choice for super-resolution. With SIM, patterned illumination is used to enhance spatial frequency information, which is usually discarded. Some SIM microscopes use a physical diffraction grating to create this pattern, but more modern techniques use sinusoidal wave patterns coming from within the actual laser light source. Regardless of the method for illumination, this pattern is then rotated and recorded 9–15 times, creating interference patterns that are then run through Fourier space to determine their precise location.
Because only 9–15 frames have to be acquired, this method can be preferred over localization microscopy for live samples. Still, it is a limitation on speed. SIM provides more options for multichannel imaging than localization microscopy. Photoswitchable fluorophores often need a separate wavelength to activate, which limits the number of fluorophores that can fit in a single specimen. That is not the case for SIM. However, SIM doesn’t achieve the same level of resolution that localization microscopy can. Practically though, most users find this suitable for live cells because of the complications associated with higher resolutions.
Stimulated emission depletion (STED) uses a single, diffraction-limited laser beam that scans across a sample in the same fashion as a laser scanning confocal. However, STED differs in that another ring-shaped laser beam in a different wavelength is also scanned across the sample to create a spectrally separate image. This second ring-shaped beam depletes the outer edge of the excitation area, so when this is subtracted from the original image, only a small portion of the original diffraction-limited beam remains visible. STED can be very high-resolution but it is important that these two laser beams be in alignment with one another, which can be difficult to perform practically in the lab.
Newer technologies for STED are making it much friendlier for live cells, such as live-cell STED with reduction of state transition cycles (RESCue STED). RESCue STED samples at a slower rate for areas where no signal is detected, which preserves the health of the samples overall, but the requirement for two laser beams per channel can be problematic since it limits the number of fluorophores that can fit into a single sample. STED is also unique in that the resolution increases as the intensity of the depletion laser intensity increases, which can be balanced to improve cell health.

Figure 2. Adding to the incredible value of HeLa cells, super-resolution can be used to image intricacies working within the cell. In this image, you can see DNA (green) and mitochondria (red) captured by the Olympus SD-OSR. (Image courtesy of Farid Jalali, Olympus Canada.)
Working with live cells
Choosing a super-resolution method, however, is only part of the consideration. The most important element to remember when working with live cells is that they must remain alive and healthy at the end of the experiment. If cells do not survive, the cause must be identified.
To keep cells healthy, the amount of light they are exposed to must be minimized. Like humans, UV light is damaging to live samples and should be avoided whenever possible. Researchers should take care to use fluorescent proteins with high quantum yields, which measures how effectively a fluorophore converts light from one wavelength to another. If this is low, a lot of light will be exposed but won’t produce much in return.
Short exposure times are preferred, and techniques to further shorten exposure times such as binning should be investigated. Again users should take caution: a 1 ms exposure with 100% laser power is not always better for the samples. Cells should also be allowed time to recover between exposures and proper imaging software should be able to accommodate differing intervals for time lapse experiments. This will allow the user to ramp up exposure during critical periods without sacrificing cell health.
Some techniques are more live cell friendly than others, like SIM. Researchers should stay current with literature regarding super-resolution, as processing algorithms are constantly improving to extract more out of samples without additional exposure to light. SIM processing, for example, can be applied to specific confocal images to achieve similar levels of resolution with fewer frames. Even localization microscopy is evolving, with high-density processing algorithms that also reduce the number of frames needed to form a super-resolution image.
Cell health is the most important factor in live-cell experiments, but there are other ways to improve the results from super-resolution experiments.

Figure 3. Actin filaments, like those shown here, make up complex networks that play important roles in cell shape and mitosis. Image captured on the Olympus SD-OSR. (Image courtesy of Farid Jalali, Olympus Canada.)
Thinking outside the box: other methods for improving results
One metric for image quality is the ratio between the signal and that which surrounds it. One of the biggest factors that hurts signal ratio is what the sample is sitting in. Super-resolution should always be performed in coverslip, glass-bottom dishes, as close to the objective as possible.
Plastic is a poor choice for super-resolution as it can autofluoresce, which is a major area of concern for live samples. Media components like phenol red and FBS can also be subject to this. Often, separate imaging media are stored without these items for use during microscopy experiments. Signal ratios can also be improved by increasing the efficiency of collecting photons through different cameras and detectors.
Outside of the microscope itself, environmental control is also critical. Optimal temperature, humidity, and gas conditions must be maintained, which is now easier than ever to accomplish on a microscope that can be adjusted as needed. This is not just important for the samples; all imaging materials, including immersion oil, should be at the same temperature. Changes can cause distortion along the optical path.
Extracellular matrix or dish coating is sometimes necessary. There are objectives available with higher working distances that can accommodate this depth but doing so comes with some risk. Each time light moves through a material with a different refractive index, spherical aberrations occur that hurt the final resolution of the image. Silicone oil objectives can be used to match the refractive index of live cells specifically, enabling better images from depths of up to 300 um. Their high numerical aperture makes them perfect for live-cell experiments since it allows as many photons as possible to be collected and sent to the detector.
To conclude
As the super-resolution microscopy industry evolves, many factors are changing alongside it, yet one thing has remained constant: a samples’ health is paramount in live cell imaging. This is true not just for super-resolution but all live cell microscopy experiments. It is important to determine the super-resolution method prior to beginning an experiment, as some methods will dictate what fluorescent proteins should be used. It is also critically important to minimize photodamage of samples for all aspects of the experiment. A major part of keeping samples alive is optimizing the microscopy system to best accommodate living samples.