It takes two: immunotherapy and cancer dance an intricate tango
Cancer is crafty. Researchers are too. And they’ve just found one reason that some cancers evade novel immunotherapies.
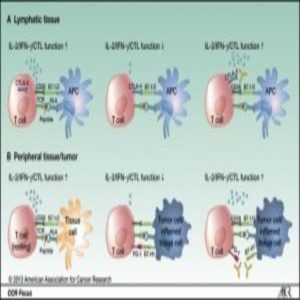
Cytotoxic lymphocyte-Associated Protein 4 and PD-1/L1: Immune checkpoint pathways that can be exploited by immunotherapy drugs to reactivate the immune system against malignant cells.
The interplay between cancer and the immune system is like a Shakespearean drama. There’s a large cast of actors, lots of spying, some double crossing, and a death at the end. Researchers seeking to enlist the help of the immune system to fight cancer have focused on surface proteins and immune checkpoints. New research, however, suggests that a bit player that only briefly occupies the spotlight, interferon gamma, has much to do about everything when it comes to this anticancer strategy.
Normally, when T-lymphocytes recognize antigens expressed on the surface of cancer cells, they generate the cytokine interferon gamma, which mediates the responses of multiple anticancer genes, including the expression of programmed death 1 (PD-1).
But cancer cells are a wily lot; they evade T-cell recognition by producing programmed death ligand 1 (PD-L1), which binds to the PD-1 receptor to signal “all-clear.” The T-cell then loses sight of its target. In this way, cancer cells evade immune recognition and are free to grow, multiply, and metastasize.
The current rising star of cancer treatment is a class of drugs called immunotherapies. Agents like nivolumab and ipilimumab target PD-1, PD-L1, or other tumor cell recognition molecules like Cytotoxic T-Lymphocyte-associated protein 4, which all act as immune checkpoints. Cancer cells interact with these checkpoints to downregulate immune activity. Immunotherapies work to reactivate these checkpoints, to pull the wool from the immune system’s eyes, as it were.
“The excitement for checkpoint blockade immunotherapy is based on durable responses for patients with advanced cancer who exhausted many options for their treatment,” said Daniel Shin, an oncology researcher at University California, Los Angeles.
Shin and his team began studying the mechanics of PD-L1 expression in cancer cells in 2013, using a large panel of previously established melanoma cell lines. They noticed that certain lines did not express PD-1 when exposed to increasing amounts of interferon. Shin subsequently found that mutations that inactivate certain genes appeared to prevent a cellular response to interferon gamma.
Shin and his team analyzed data from clinical trials of anti-PD-1 drugs and discovered that although patients with high mutational loads in biopsied tumor cells usually responded well to PD-1 blockade therapy, there always seemed to a be a small contingent of subjects that did not respond at all.
“Initially, we doubted our findings,” said Shin. But the research bore out. Ostensibly good candidates for immunotherapy simply did not respond to the drugs.
“Our findings are now corroborated by other groups of investigators who reported interferon gamma receptor mutations as resistance mechanisms of ipilimumab,” Shin said.
Shin and his team hypothesized that loss of function mutations in the genes JAK1 and JAK2, which mediate the receptors for interferon gamma, provided the genetic mechanism by which certain patients present primary resistance to immunotherapeutic agents.
Shin performed genetic analysis of tumors from melanoma and colon cancer patients who had a high mutational load but did not respond to PD-1 blockade or PD-L1 inhibition therapy. They discovered that loss of JAK1/2 function correlated with a lack of response to immunotherapy. Then Shin and his team compared wild-type melanoma cell lines against JAK1/2 mutated cell lines to assess their response to interferon gamma. As they predicted, the mutated cell lines did not increase their expression of PD-L1, rendering them invulnerable to anti-PD-L1 therapy.
Shin and his team wanted to establish a causal relationship between JAK1/2 and interferon gamma, so they employed a lentivirus to introduce wild-type JAK1 back into a mutated cell line. The reintroduced gene did, in fact, restore the response to interferon gamma. The cells produced PD-L1 at an increased rate, rendering them susceptible to anti-PD-1 or PD-L1 therapies.
The results are important to ensure a proper fit between patient and treatment regimen. “We think that checkpoint blockade immunotherapy [drugs that inhibit cancer cells’ ability to produce PD-1/L1] would not be effective for those who harbor JAK1/2 loss of function mutations,” said Shin.
Researchers are also investigating workaround tactics for resistant patients.“We are currently conducting studies by using mouse model to test various strategies for how we can overcome the JAK mutations,” said Shin.
“The field is moving so fast with global research efforts,” he said. “I anticipate that we will see steady advancements.