How does diabetes spread? Mad cow disease could be the answer
Mad cow disease made headlines years ago, becoming infamous for its unique mode of transmission. Now, evidence is mounting that diseases as common as diabetes and Parkinson’s disease are spread by similar mechanisms.

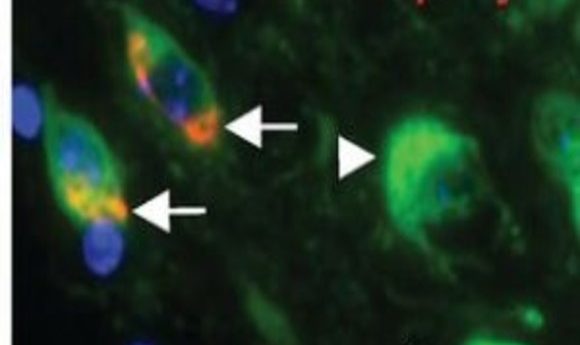
Colocalization of syn inclusions with dopamine neurons (5).
Mad cow disease became a household worry during the late 80s and throughout the 90s. As the disease ravaged cattle throughout Great Britain, it raised alarm for meat-eaters who feared contracting the disease from contaminated animal products. The epidemic introduced the world to prions—proteinaceous infectious particles—a term coined by Stanley Prusiner at the University of California, San Francisco in 1982 when he identified them as the infectious agents responsible for bovine spongiform encephalopathy (mad cow disease), scrapie in sheep, and Creutzfeldt-Jakob disease in humans. This discovery earned him the Nobel Prize in physiology or medicine in 1997.
While prion diseases can spread in patterns similar to those of bacterial, viral, fungal, or parasitic infections, they are unique in that no nucleic acids are involved—the protein alone conveys the disease. Prions can fold into multiple structures, at least one of which is pathogenic. This pathogenic form serves as a template to refold healthy cellular proteins into an amyloid form, consisting of tightly packed beta sheets that aggregate together into fibrils. Not only do cells suffer the loss of the healthy protein functions, but the aggregates damage cells and resist normal cellular protein disposal processes.
The idea that a lone protein might transmit disease was once controversial but has gained traction over the years with accumulating experimental evidence. Recent studies actually indicated that this mode of transmission extends beyond the previously recognized prion diseases. Alzheimer’s disease, Parkinson’s disease (PD), multiple system atrophy, AA amyloidosis, and diabetes all employ prion-like strategies for disease progression.
“I don’t think that most researchers in the general amyloid field completely understood the potential impact on other protein aggregation diseases,” remarked Per Westermark from Uppsala University. “Prusiner nowadays calls all amyloid diseases prion diseases.”
Aggregation Aggravation
Westermark’s team first began exploring the spread of amyloid deposits in the early 90s while working with mouse models of AA amyloidosis, a disease that occasionally follows other inflammatory illnesses. During inflammation, the liver produces high levels of serum amyloid A protein (SAA), which aggregates on rare occasions. These aggregates deposit throughout the body, causing significant health problems when they lodge in the kidneys.
Researchers working in this field noticed that mice developed amyloidosis quicker following exposure to tissue extracts from other amyloidotic mice. While many scientists attempted to isolate the “amyloid enhancing factor” in these tissue extracts, its identity remained a mystery.
“Our idea came from knowledge about prion diseases and that all amyloid shares a β-sheet structure while the parent protein may be most α-helical. Therefore, we tried synthetic fibrils from short peptides and found that these exert similar amyloidosis-accelerating effects, although weaker than an extract from native amyloid deposits,” Westermark recalled (1).
From there, the team went back to the tissue extracts used to accelerate amyloidosis in their mouse models to look for a component that served the same function as the synthetic fibrils. In 2002, they reported that amyloid fibrils result from nucleation by misfolded forms of SAA. The misfolded protein was very potent in directing aggregation of high levels of SAA circulating during inflammation, so potent in fact, that injection wasn’t required; SAA fibrils could exert their effects even when mice ate the misfolded protein (2).
“From these studies, it was natural to ask if also other amyloid diseases may be transferable,” Westermark said.
Prion Propagation
Across the ocean and nearly a decade later, Kelvin Luk joined the laboratory of Virginia Lee at the University of Pennsylvania and began trying to form Lewy body inclusions—protein aggregates seen in PD—in cultured cells. Luk was familiar with studies showing that prions propagated when directly introduced into yeast and that alpha-synuclein (α-syn; the main component of Lewy bodies) misfolding accelerated in the presence of aggregates. “Our idea was to combine these two methods and it turned out quite successfully,” Luk said.
With their in vitro model in place, the team showed that introducing α-syn fibrils to cells overexpressing healthy α-syn recruited protein into insoluble, hyperphosphorylated, ubiquitinated aggregates—Lewy bodies (3). Overexpressing α-syn wasn’t necessary for inducing aggregation, and aggregation led to neuron death (4).
At the time, PD researchers characterized the disease by looking for α-syn inclusions and a loss of midbrain dopamine neurons, but a cause and effect relationship hadn’t been established. To close this gap, Luk’s team turned to mouse models, and showed that a single inoculation of α-syn fibrils led to cell-to-cell transmission of pathologic α-syn, Lewy body accumulation, and loss of dopamine neurons. Importantly, aggregates propagated along connected neurons, showing up in cells far from the site of injection (5,6).
“The observations provided direct experimental evidence for pathological α-syn propagation in vivo and encouraged the field to really embrace the idea of pathological spread in PD,” Luk said. “The fact that the models we described required only recombinant protein and widely available mouse lines (or non-transgenic mice) made it easy for labs to adopt this approach for both basic research and drug discovery.”
More recently, researchers have gathered additional evidence for prion-like transmission in PD, as well as several other diseases including type 2 diabetes (read more on How Type 2 Diabetes is Similar to Mad Cow Disease), yet exactly how the misfolded protein induces amyloid formation remains a mystery.
“This is still not a very well-defined process and is hotly debated,” Luk said. “Neurons or other cells containing pathological α-syn likely release some of this into the extracellular space, maybe through a combination of exocytosis and cell death, which is then taken up by neighboring cells. It is quite possible that other intermediary cells are also involved in this process.”
Cause for Concern
In humans, 31 different proteins have the potential to form amyloids, 14 of which associate with systemic diseases. To initiate disease, the amyloid fibril precursor need only express in a tissue, circulate in the blood, and deposit in an organ. In fact, by the age of 50, most people likely have at least one type of amyloid deposit somewhere in their body. Assuming a high enough concentration of native protein for conversion, the rate of prion aggregation can grow exponentially. Given the quick spread of mad cow disease and the fact that mice only needed to eat something contaminated with seed protein to develop Lewy bodies, could diseases such as diabetes or PD be contagious in humans?
“There is still no definite proof for such transmission [in humans], but there are some indirect indications. In animal experimental systems, there are proofs, however,” Westermark said. Aside from mice, amyloids can pass between individual cheetahs, foxes, and gulls, probably through contact with feces containing amyloid fibrils. “Transmission is most likely possible for all amyloid diseases, but we are probably not exposed to many of the amyloids in a way that can cause transmission. Or we may not have the substrate in concentration enough to cause seeding.”
Westermark is most interested in exploring the idea of cross-seeding—whether one amyloid type can promote formation of another. “We have some experimental indications that this may be the case in islet amyloid polypeptide (IAPP, the amyloid protein in type 2 diabetes) interaction with Aβ (the protein in Alzheimer’s disease,” he said. “Cross-seeding is not only possible between amyloid proteins but also between natural protein polymers in nature and amyloid proteins. For example, some bacterial and fungal proteins adopt amyloid-like conformations and may interact with or cross-seed human amyloid proteins.”
Westermark is also interested in determining how amyloid deposits spread in the body, a goal shared by Luk, who plans to focus his next studies on how pathology affects specific regions of the nervous system.
“Although α-syn misfolding is clearly important, we also know very little about α-syn itself,” Luk said. Learning the basic biology of the protein should significantly advance understanding of why the aberrant protein becomes so damaging, lending insights into the role protein loss plays as proteins misfold, as well as how aggregates spread from cell to cell. “I think this strategy will eventually pay rich dividends.”